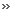RESULTS
Purification and properties of rubber oxygenase (RoxA).
Xanthomonas sp. produces clearing zones during growth on opaque
mineral salts agar with latex as a carbon source. Apparently,
Xanthomonas sp. secretes an activity that is able to degrade
latex to water-soluble products (Fig. 1A). In order to
establish an in vitro assay for latex degradation and to analyze the
latex-degrading activity, Xanthomonas sp. was grown on mineral
salts medium with purified latex. After 7 to 9 days of incubation the
latex was visibly degraded and/or coagulated. The culture fluid was
separated from the cells and remaining latex particles by successive
centrifugation and filtration (pore size, 0.2 �m). Macromolecular
components were concentrated about 20-fold by ultrafiltration
(30-kDa cutoff). When aliquots of fresh latex were added to the
cell-free concentrated culture fluid and incubated at 30�C, latex was
again visibly coagulated or degraded within 2 days; the control, with
buffer instead of culture fluid, remained milky. When the same
experiment was repeated with either the flowthrough from the 30-kDa
filtration step or the concentrated culture fluid that had been
heated to 95�C for 10 min before latex was added, no coagulation or
degradation was observed. We concluded that the compound responsible
for coagulation or degradation of the latex is a heat-sensitive
macromolecule, presumably an enzyme. It was noticed that the
concentrate had a light reddish color that did not appear to the same
extent in glucose-grown cultures. The concentrate had a single
absorption maximum around 406 nm (the range from 400 to 600 nm was
tested) that could be shifted upon reduction to
 418 and
550 nm (data not shown). These
results are characteristic for heme-containing proteins, and we
speculated that a heme-containing protein was responsible for latex
degradation. Using 2 liters of cell-free culture fluid of NR-grown
Xanthomonas sp. as the starting material, we were able to purify
a protein (apparent molecular mass after sodium dodecyl sulfate [SDS]-polyacrylamide
gel electrophoresis, 65 � 7 kDa) to apparent electrophoretic
homogeneity by diafiltration and subsequent chromatography on Q-Sepharose
and MonoP (chromatofocusing). The purified 65-kDa protein showed
the same strong absorption at 406 nm as the concentrated culture
fluid (Fig. 1B and C); the oxidized protein had
absorption bands at 280 nm (
418 and
550 nm (data not shown). These
results are characteristic for heme-containing proteins, and we
speculated that a heme-containing protein was responsible for latex
degradation. Using 2 liters of cell-free culture fluid of NR-grown
Xanthomonas sp. as the starting material, we were able to purify
a protein (apparent molecular mass after sodium dodecyl sulfate [SDS]-polyacrylamide
gel electrophoresis, 65 � 7 kDa) to apparent electrophoretic
homogeneity by diafiltration and subsequent chromatography on Q-Sepharose
and MonoP (chromatofocusing). The purified 65-kDa protein showed
the same strong absorption at 406 nm as the concentrated culture
fluid (Fig. 1B and C); the oxidized protein had
absorption bands at 280 nm ( band), 356 nm, and 406 nm (Soret band) and weaker and broader bands
at 532 nm (� band), 560 to 565 nm (
band), 356 nm, and 406 nm (Soret band) and weaker and broader bands
at 532 nm (� band), 560 to 565 nm ( band), and 672 nm. A molar extinction coefficient of 1.8 x 105
M�1 cm�1 was determined for the absorption at
406 nm, which was similar to the coefficients of diheme enzymes .
The
band), and 672 nm. A molar extinction coefficient of 1.8 x 105
M�1 cm�1 was determined for the absorption at
406 nm, which was similar to the coefficients of diheme enzymes .
The  406/280
value, which reflected the purity and spectral characteristics of
RoxA, was 1.17 in 20 mM phosphate buffer (pH 6.8). When the purified
65-kDa protein was reduced by Na2S2O4,
absorption, bands appeared at 418 nm (Soret band), 522 nm (�
band), and 549 and 553 nm (both
bands), which corresponded to a heme-pyridine complex. These data are
indicative of a hemoprotein belonging to the cytochrome c
group . Addition of synthetic oligo(1,4-cis-isoprene)
to purified RoxA resulted in a shift of the Soret band from 406 to
409 nm, indicating that the substrate binds to the enzyme at the heme
site(s). The optical spectra of purified RoxA did not exhibit an
absorption band at 695 nm which would be expected for a heme iron-methionine
bond.
406/280
value, which reflected the purity and spectral characteristics of
RoxA, was 1.17 in 20 mM phosphate buffer (pH 6.8). When the purified
65-kDa protein was reduced by Na2S2O4,
absorption, bands appeared at 418 nm (Soret band), 522 nm (�
band), and 549 and 553 nm (both
bands), which corresponded to a heme-pyridine complex. These data are
indicative of a hemoprotein belonging to the cytochrome c
group . Addition of synthetic oligo(1,4-cis-isoprene)
to purified RoxA resulted in a shift of the Soret band from 406 to
409 nm, indicating that the substrate binds to the enzyme at the heme
site(s). The optical spectra of purified RoxA did not exhibit an
absorption band at 695 nm which would be expected for a heme iron-methionine
bond.
View larger version (44K):
|
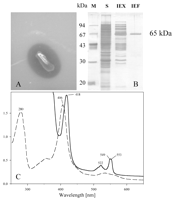
FIG. 1. (A) Clearing zone formation for
Xanthomonas sp. after 4 days of incubation at 30�C on opaque latex
agar. (B) SDS-polyacrylamide gel electrophoresis of the 65-kDa protein (RoxA)
at various stages of purification. Lane M, marker proteins; lane S,
concentrated cell-free culture fluid; lane IEX, pool after ion-exchange
chromatography on Q-Sepharose; lane IEF, pool after isoelectric focusing
on MonoP. (C) Spectra of purified RoxA before (dashed line) and after
(solid line) reduction by dithionite. The numbers indicate the observed
maxima of the spectra.
|
|
A trypsin fingerprint analysis of the isolated 65-kDa protein was
performed, and the masses of the peptides generated were determined
by matrix-assisted laser desorption ionization�time of flight
analysis (data not shown). The values obtained for six randomly
isolated peptides were in agreement with the values obtained for an
in silico trypsin digest of a gene product encoded by a recently
cloned gene of Xanthomonas sp. . The cloned
gene was assumed to be involved in rubber degradation, but the
biochemical function of the protein was not known. Determination of
the amino acid sequences of these six peptides confirmed that the
65-kDa protein was identical to the gene product mentioned above
(peptide 1, NH2-YGLYPAPFR; peptide 2, NH2-TTPITALGNLLPLPWSTGR;
peptide 3, NH2-GLEDEFEDINNFLISLSPATYPK; peptide 4, NH2-GVAAVVTPIETIR;
peptide 5, NH2-AWNSGWWAYNNLSPSWTGYPSDNIVASELR; peptide 6, NH2-WALIEYIK).
The deduced amino acid sequence of the cloned gene contained
two heme-binding motifs (CXXCH) typical for covalently bound heme.
Calculation of the heme content of the purified 65-kDa protein by
using the molar absorption coefficient of heme-cytochromes ( ,
29.1 mM�1 cm�1 at 551 nm [5]) and three
independent methods for protein determination revealed heme and
protein concentrations of 16.7 and 9 �M, respectively. These values
correspond to a heme content of 1.9 mol of heme per mol of the
65-kDa protein and are in good agreement with the gene sequence
analysis that predicted two heme-binding sites. An apparent molecular
mass of 54 kDa for the purified native protein was determined by gel
filtration (Sephadex G-200). Apparently, the protein has a monomeric
subunit structure. In most experiments, the SDS-denatured (reduced)
protein migrated at values corresponding to an apparent molecular
mass of 55 to 65 kDa, which were significantly lower than the
theoretical molecular mass of the mature protein (72.9 kDa) deduced
from the gene sequence. The reason for the discrepancy in apparent
molecular masses is not known. To test whether the purified protein
was responsible for the observed latex-coagulating and -degrading
activity, it was added to diluted latex and incubated at 30�C. After
incubation for 24 h, controls without enzyme or with boiled enzyme
were not changed, but clearing and coagulation of the latex were
visible upon incubation with the active enzyme, confirming that the
purified 65-kDa protein was responsible for the latex-degrading and
-coagulating activity.
,
29.1 mM�1 cm�1 at 551 nm [5]) and three
independent methods for protein determination revealed heme and
protein concentrations of 16.7 and 9 �M, respectively. These values
correspond to a heme content of 1.9 mol of heme per mol of the
65-kDa protein and are in good agreement with the gene sequence
analysis that predicted two heme-binding sites. An apparent molecular
mass of 54 kDa for the purified native protein was determined by gel
filtration (Sephadex G-200). Apparently, the protein has a monomeric
subunit structure. In most experiments, the SDS-denatured (reduced)
protein migrated at values corresponding to an apparent molecular
mass of 55 to 65 kDa, which were significantly lower than the
theoretical molecular mass of the mature protein (72.9 kDa) deduced
from the gene sequence. The reason for the discrepancy in apparent
molecular masses is not known. To test whether the purified protein
was responsible for the observed latex-coagulating and -degrading
activity, it was added to diluted latex and incubated at 30�C. After
incubation for 24 h, controls without enzyme or with boiled enzyme
were not changed, but clearing and coagulation of the latex were
visible upon incubation with the active enzyme, confirming that the
purified 65-kDa protein was responsible for the latex-degrading and
-coagulating activity.
Isolation and identification of the main cleavage
product of enzymatic rubber degradation.
In order to determine whether the purified protein cleaved the carbon
backbone of the polymer or whether it only affected the integrity of
the latex emulsion (i.e., the diameter-to-volume ratio of the latex
particles), we investigated the presence of low-molecular-mass
degradation products in the cleared and coagulated latex. Latex was
incubated with the purified 65-kDa protein for several hours at 30�C
and subsequently extracted with ethyl acetate; the extracts were
analyzed by two-dimensional TLC. One major dark spot became visible
when the TLC plate was developed with anisaldehyde, and this spot was
absent in controls with either no enzyme or heat-inactivated enzyme
(Fig. 2). A few minor low-intensity spots of
different color also appeared in some experiments. HPLC analysis of
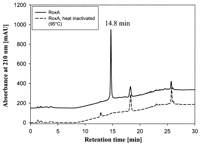
the same ethyl acetate extract revealed only one major peak (at a
retention time of 14.8 min) that was missing in control experiments
(Fig. 3). The identity of the 14.8-min HPLC
fraction that produced the large spot in TLC analysis was established
by collecting the corresponding HPLC fraction; subsequent
two-dimensional TLC analysis revealed only one spot with the same
Rf value (data not shown).

View larger version (66K):
|
FIG. 2. Two-dimensional thin-layer
chromatography of degradation products produced from latex by purified
RoxA. Latex was allowed to react with purified RoxA before products were
extracted with ethyl acetate, dried, and resolved with methanol.
Aliquots were spotted onto TLC plates, separated with benzene-acetone
(20:1) in the first dimension and with chloroform-methanol (9:1; 90�) in
the second dimension, and developed with anisaldehyde-H2SO4
at 100�C.
|
|
View larger version (20K):
|
FIG. 3. Separation of polyisoprene
degradation products by HPLC. Latex was incubated with purified RoxA for
3 h at 40�C and pH 7. Ethyl acetate-extracted products were loaded on a
C8 reverse-phase HPLC column as described in Materials and
Methods. Heat-inactivated RoxA served as a negative control. mAU, milli
absorbance units.
|
|
The compound which appeared as large dark spot in TLC and as the only
prominent peak at 14.8 min in the HPLC analysis thus apparently
represented the principal enzymatic rubber degradation product. In a
coupled HPLC-MS analysis in the negative ESI mode, the mass spectrum
of the 14.8-min fraction showed an [M-H]� peak at m/z 235
(Fig. 4). When the total ion chromatogram from HPLC
was analyzed for additional signals at m/z values that
differed from m/z 235 by n isoprene units (i.e.,
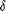 m/z 68), additional
[M-H]� peaks were detected in the m/z 167, 303, 371, and
439 ion chromatograms (Table 1). The levels of these
minor metabolites apparently were below the detection limit of the UV
diode array in the HPLC analysis. They represent a series of
homologous degradation products with one isoprene unit less or one to
three isoprene units more than the major metabolite (236 Da).
m/z 68), additional
[M-H]� peaks were detected in the m/z 167, 303, 371, and
439 ion chromatograms (Table 1). The levels of these
minor metabolites apparently were below the detection limit of the UV
diode array in the HPLC analysis. They represent a series of
homologous degradation products with one isoprene unit less or one to
three isoprene units more than the major metabolite (236 Da).
View larger version (9K):
|
FIG. 4. HPLC-ESI-MS analysis of latex
degradation products. Latex degradation products were prepared and
separated by HPLC as described in the legend to Fig. 3,
and the eluate was monitored by negative ESI-MS. The graph shows the
average mass spectrum summed across the 14.8-min peak (seven scans). The
relative intensity of the (M+1)-13C isotope peak (m/z
236) corresponds to a C15 molecular formula.
|
|
The positive ESI mass spectrum of the major metabolite showed an [MH]+
peak at m/z 237, as well as the regular adduct ions [M+Na]+and
[M+K]+ and the corresponding cluster ions with methanol (m/z
259, 275, 291, and 307). Two intense peaks at m/z 219 and 201
represented the loss of one or two water molecules from the
quasimolecular ion MH+, which definitively established the
incorporation of two oxygen atoms in the metabolite (data not shown).
When the isolated 236-Da compound was reacted with
dinitrophenylhydrazine, a yellow product was obtained, indicating the
presence of carbonyl functions in the molecule. Additional structural
information was obtained by 1H-NMR spectroscopy of the
isolated compound; the individual resonance signals are shown in
Table 2 together with the first-order multiplicity
and the corresponding assignments. For the resonance at the lowest
field, the chemical shift ( 9.78 ppm), relative intensity 1H, and vicinal coupling constant
to the -methylene protons (CH2,
1.75 Hz) are characteristic of an aldehyde proton. The sharp singlet
at 2.14 ppm (relative intensity 3H), on the other hand, is indicative
of a nonconjugated acetyl group. Thus, the two ends of the main
metabolite can be definitively identified as CHO-CH2
9.78 ppm), relative intensity 1H, and vicinal coupling constant
to the -methylene protons (CH2,
1.75 Hz) are characteristic of an aldehyde proton. The sharp singlet
at 2.14 ppm (relative intensity 3H), on the other hand, is indicative
of a nonconjugated acetyl group. Thus, the two ends of the main
metabolite can be definitively identified as CHO-CH2 and
CH2-CO-CH3,
with a combined mass contribution of 100 Da. Since the overall
molecular mass is 236 Da, this leaves 136 Da for the core of the
metabolite, corresponding to two isoprene moieties [&0807;CH2-C(CH3)
and
CH2-CO-CH3,
with a combined mass contribution of 100 Da. Since the overall
molecular mass is 236 Da, this leaves 136 Da for the core of the
metabolite, corresponding to two isoprene moieties [&0807;CH2-C(CH3) CH-CH2;
68 Da each]. The two expected olefinic proton signals for the
main metabolite (n = 2) (Fig. 5) were observed at
5.17 and 5.13 ppm. We
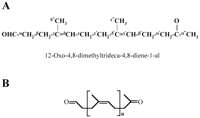
concluded that 12-oxo-4,8-dimethyltrideca-4,8-diene-1-al (ODTD) (m/z
236) is the formula of the isolated degradation product with a
retention time of 14.8 min in HPLC analysis. The experiments
described above showed for the first time that in vitro a single
enzyme is capable of cleaving the carbon backbone of rubber, yielding
12-oxo-4,8-dimethyltrideca-4,8-diene-1-al as the major degradation
product. The purified protein and its corresponding gene were
designated rubber oxygenase A (RoxA) and roxA, respectively.
CH-CH2;
68 Da each]. The two expected olefinic proton signals for the
main metabolite (n = 2) (Fig. 5) were observed at
5.17 and 5.13 ppm. We
concluded that 12-oxo-4,8-dimethyltrideca-4,8-diene-1-al (ODTD) (m/z
236) is the formula of the isolated degradation product with a
retention time of 14.8 min in HPLC analysis. The experiments
described above showed for the first time that in vitro a single
enzyme is capable of cleaving the carbon backbone of rubber, yielding
12-oxo-4,8-dimethyltrideca-4,8-diene-1-al as the major degradation
product. The purified protein and its corresponding gene were
designated rubber oxygenase A (RoxA) and roxA, respectively.
View larger version (11K):
|
FIG. 5. Proposed structure of the latex
degradation products produced by RoxA. (A) Molecular structure of the
main metabolite at 14.8 min in the HPLC analysis
(12-oxo-4,8-dimethyltrideca-4,8-diene-1-al), including the assignments
of the 1H-NMR signals (Table 2). (B)
General structure of the unequivocally characterized major and minor
degradation products, with n = 1 to 5.
|
|
Biochemical characterization of the NR cleavage reaction with rubber
oxygenase (RoxA).
An assay for determination of RoxA activity was developed as follows.
The standard assay was performed (see Materials and Methods), and the
ethyl acetate-extracted products were separated by HPLC. The RoxA
activity was calculated from the peak area at 14.8 min. At low
concentrations of RoxA (0.01 to 2 �g/ml of assay mixture) a linear
relationship between the area of the 14.8-min peak and the RoxA
concentration was found (data not shown). The optimum pH and
temperature of purified RoxA were determined to be around pH 7 and
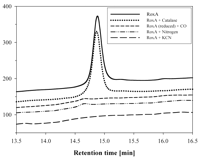
40�C, respectively (Fig. 6). The reaction was
strictly dependent on the presence of oxygen, and no rubber
degradation product was detected in a nitrogen atmosphere. Addition
of potassium cyanide (20 mM) to the assay mixture completely
inhibited the reaction. Carbon monoxide also inhibited the reaction
if RoxA had been reduced by dithionite before the assay was started
in the presence of oxygen (Fig. 7). The results
described above are in agreement with the presence of an essential
heme(s) in the enzyme. Addition of catalase at various concentrations
to the assay system did not affect the reaction at all. Peroxidase
activity was not detected even after a prolonged incubation time or
if high concentrations of RoxA were used, while a positive control
(horseradish peroxidase) reacted within seconds. Alkylation agents,
such as N-ethylmaleimide and p-chlorobenzoate, and
chelators (bispyridyl, tiron) partially inhibited the reaction.
However, EDTA had no significant effect on the activity. Reducing
agents and all of the detergents tested (except cholate) strongly
inhibited the reaction (Table 3).
View larger version (19K):
|
FIG. 7. Dependence of RoxA activity on
oxygen and effects of cyanide, carbon monoxide, and catalase. One
microgram of purified RoxA was allowed to react with latex in the
presence of different compounds. Ethyl acetate extracts were analyzed to
determine the area of the peak at 14.8 min by HPLC. Treatments: control
(air with 21% oxygen) (RoxA); catalase (1 mM); carbon monoxide (first
the atmosphere was replaced by N2 before N2 was
replaced by CO, and the assay was subsequently performed under normal
air with 21% oxygen); air replaced
by N2; addition of 20 mM potassium cyanide.
|
|
RoxA appeared to be specific for oligomers and polymers of 1,4-isoprene.
Natural latex and chemosynthetic poly(cis-1,4-isoprene) were
significantly cleaved by purified RoxA. When a mixture of chemosynthetic
rubber [oligo(cis-1,4-isoprene) with 5 to 15 isoprene units
(Mw, 790; Mn, 707; Mw/Mn,
1.12)] was incubated with RoxA, a peak at 14.8 min appeared as a
degradation product in HPLC, and the composition of the remaining
oligomer mixture was shifted to low-molecular-weight products (data
not shown). Oligomers of trans-1,4-isoprene, such as squalene,
were hardly cleaved by RoxA, and only small amounts of
carbonyl-containing compounds, which were slightly above the
detection limit, were detected. When toluene was tested as a
substrate, only background activity was obtained. Benzene did not
react at all.





 , 0.2 M bis-Tris-HCl;
, 0.2 M bis-Tris-HCl;
 , 0.2 M
Tris-HCl;
, 0.2 M
Tris-HCl;  , 0.2 M
ethanolamine-HCl.
, 0.2 M
ethanolamine-HCl.