MATERIALS AND METHODS
Bacteria, media, and culture conditions.
Xanthomonas sp. was grown in nutrient broth or in a mineral
salts medium described by Tsuchii and Takeda with 0.5%
glucose or 0.2% purified rubber latex at 30�C. Latex cultures
also contained 0.002% Tween 80 and sometimes contained 0.05% yeast
extract. Solid media contained 1.5% agar. Latex agar was prepared by
the overlay technique; a bottom layer ( 30
ml) of mineral salts agar in a petri disk was overlaid with the same
agar supplemented with 0.2% purified latex from H. brasiliensis
(percentage of solid rubber) with or without 0.05% yeast extract,
which resulted in an opaque overlay. Colonies of Xanthomonas
sp. produced translucent clearing zones upon incubation at 30�C
within 2 to 4 days, indicating utilization of the latex.
30
ml) of mineral salts agar in a petri disk was overlaid with the same
agar supplemented with 0.2% purified latex from H. brasiliensis
(percentage of solid rubber) with or without 0.05% yeast extract,
which resulted in an opaque overlay. Colonies of Xanthomonas
sp. produced translucent clearing zones upon incubation at 30�C
within 2 to 4 days, indicating utilization of the latex.
Rubbers.
Rubber latex was prepared from freshly tapped H. brasiliensis.
Crude latex contains approximately 35% rubber and 1 to 1.5% proteins.
Latex was purified from soluble proteins by repeated (three times)
centrifugation and washing with 0.002% Tween 80. The top layer
(cream) from each centrifugation step was used for the next
centrifugation step, while the bottom fractions were discarded. Latex
was heat sterilized and stored at 4�C. Purified latex was a gift from
the Rubber Research Institute of Malaysia.
Purification of rubber oxygenase (RoxA).
Rubber oxygenase was purified at 5�C by using a fast-performance
liquid chromatography system consisting of an LCC 500 controller, a
500 pump, a UV-1 monitor, a Rec-482 recorder, and an FRAC autosampler
(Pharmacia, Uppsala, Sweden). Cell-free supernatant of latex-grown
Xanthomonas sp. cells was concentrated by ultrafiltration (30-kDa
cutoff) and passed through a Q-Sepharose column (HP HR16/10;
Pharmacia) that was preequilibrated with basic buffer (20 mM
ethanolamine-HCl [pH 9.5]) at a flow rate of 1 ml min�1.
RoxA was eluted from the column with a linear gradient of 0 to 0.15 M
NaCl in basic buffer at a concentration of approximately 15 mM.
Fractions showing the characteristic absorption spectrum of RoxA were
pooled and, after desalting and changing of the buffer by
diafiltration (30-kDa cutoff), were applied to a MonoP column (HR
5/5; Pharmacia) that was preequilibrated with 20 mM
1,3-diaminopropane-HCl (pH 11.0) at a flow rate of 0.5 ml min�1.
During elution with a linear pH gradient (Pharmalyte HCl [pH 8.5]
1:60; Pharmacia) peaks with the characteristic spectrum of RoxA were
observed. These fractions were pooled and passed through a Superdex
200 column (Superdex 200 Prep-grade; Pharmacia) and eluted with 20 mM
phosphate buffer (pH 7.0).
Protein determination.
Routinely, protein determinations were performed by the method of
Bradford . For determination of the heme content, the
concentration of purified RoxA was also determined by the bicinchoninic
acid assay at 562 nm by using a commercial kit (Perbio Science,
Erembodegem, Belgium) and by determining the absorption at 280 nm
with a specific molar absorption coefficient of 153,160 M�1cm�1,
which was calculated from the amino acid composition as described
by Gill and Hippel ).
Determination of heme content.
The heme content of purified RoxA was determined by the pyridine
ferrohemochromogen test . Six hundred microliters of
purified RoxA (15 �g/ml, as determined by the bicinchoninic acid
assay and by the Gill-Hippel assay was added to an
aqueous solution of alkaline pyridine (final concentrations, 7.5 mM
NaOH and 25% pyridine; final volume, 800 �l) and reduced by
adding a few crystals of sodium dithionite. The heme content was
calculated from the absorption at 551 nm ( ,
29.1 mM�1 cm�1).
,
29.1 mM�1 cm�1).
Determination of carbonyl content.
The carbonyl content of rubber degradation products was determined
after formation of 2,4-dinitrophenyl hydrazones (in hexane) as
described by Katz and Keeney by using a molar absorbance
coefficient of 21,500 M�1 cm�1 at 338 nm.
RoxA assay and peroxidase assay.
The following conditions were used for product analysis after RoxA-catalyzed
rubber degradation by thin-layer chromatography (TLC), carbonyl
content determination, and high-performance liquid chromatography (HPLC).
The reaction mixture (total volume, 1 ml) contained 100 �l of
purified RoxA (10 to 15 �g/ml), rubber latex (4 �l of a 35%
emulsion), and bis-Tris buffer (200 mM; pH 7.0). The reaction was
carried out at 40�C for 3 or 4 h in a test tube closed with Parafilm.
The mixture was extracted with ethyl acetate or diethyl ether, dried,
dissolved in 100 to 200 �l of methanol, and then subjected to TLC,
carbonyl content determination, or HPLC analysis. Mixtures without
RoxA and with heat-inactivated RoxA (10 min, 95�C) were used as
negative controls. One unit of activity corresponded to 1 �mol of
generated carbonyl function per min. Peroxidase activity was assayed
at 510 nm as described by Mason et al. ; RoxA
was incubated in 100 mM sodium phosphate buffer (pH 7) containing 5
mM 2,4 dichlorophenol, 3.2 mM 4-aminoantipyrene, and 1 mM hydrogen
peroxide. Horseradish peroxidase was used as a positive control.
Analysis of rubber degradation products by
two-dimensional TLC.
Ethyl acetate extracts dissolved in methanol were spotted onto TLC
plates (Kieselgel 60F254; Merck & Co., Inc.), and each plate was
developed with benzene-acetone (20:1) in the first dimension and with
chloroform-methanol (9:1) in the second dimension. After evaporation
of the solvent the plates were developed with anisaldehyde-H2SO4
spray reagent.
Analysis of reaction products by HPLC and HPLC-mass
spectrometry (MS).
Degradation products were detected at 210 nm by HPLC analysis (Chromeleon
Chromatography Data Systems 4.38 equipped with a Dionex UV7Vis
detector, a UVD 170S/340S, a Dionex P 580 pump, and a Dionex Gina 50
autosampler; Dionex, Isstein, Germany) with a Grom-Sil 100 RP-8
column (125 by 4 mm; particle size, 5 �m; Grom, Herrenberg, Germany).
The mobile phase consisted of 50% (vol/vol) aqueous methanol, and
separation of the samples was carried out with a gradient to 100% (vol/vol)
methanol. Liquid chromatography mass spectra were obtained in the
negative and positive electrospray ionization (ESI) mode with an
HP1100 HPLC system (Hewlett-Packard, Waldbronn, Germany) coupled to
a VG Platform II quadrupol mass spectrometer (Micromass, Manchester,
United Kingdom). Samples were resolved with the column and mobile
phase described above.
Spectral analysis.
Proton nuclear magnetic resonance (1H-NMR) spectra were obtained
with an ARX 500 spectrometer (Bruker, Rheinstetten, Germany) at
a nominal frequency of 500.15 MHz. Samples were dissolved in CDCl3.
Chemical shifts (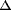 ) were
expressed in parts per million relative to tetramethylsilane as an
internal standard.
) were
expressed in parts per million relative to tetramethylsilane as an
internal standard.